
Myofibrillar myopathies (MFM) are genetic neuromuscular disease that leads to progressive muscle deterioration. They are group of skeletal and cardiac muscle disorders characterized by destruction of the muscle fiber at Z disk and accumulation of protein aggregates. The signs and symptoms of MFM may vary among individuals, depending among the genetic cause of the disease. Muscle weakness (myopathy) begins in mid-adulthood. Some patient experience first muscle weakness in the distal muscles (hand and legs) whereas some may experience in proximal muscles (muscles near the center of the body). Muscle weakness worsens over the time. Other signs and symptoms may include cardiomyopathy, arrythmia, myalgia and loss of sensation in limbs. Some people may develop cataracts (clouding of the front surface of the eyes), joint stiffness and scoliosis (abnormal curvature of the spine).
Myofibrillar myopathy was first described in 1996. Prior to coining the term MFM in 1996, these disorders were known as the term ‘desmin related myopathies’ or ‘desmin storage myopathies’.
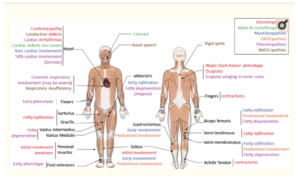
Figure- Clinical Overview of MFM alterations. Each color represent feature found in one pathology. (Source- Sabrina et al, 2017)
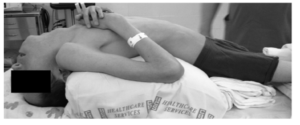
Figure- Teenager with BAG3-myopathy in natural posture due to severe scoliosis, rigid spine, and neck extension contracture. (Source- Gregory et al, 2015)
Cause of Myofibrillar Myopathies
MFM is caused by mutation in any of several genes, including desmin (DES), ab-crystallin (CRYAB), myotilin (MYOT), Z band alternatively spliced PDZ-containing protein (ZASP), filamin C (FLNC), and Bcl-2-associated athanogene-3 (BAG3), four and a half LIM domain protein 1 (FHL1) (5), plectin (PLEC) (6), and titin (TTN). So far, pathogenic mutations have been identified in 17 genes. Some of these genes are also associated with other forms of muscle diseases. These genes give instruction to our bodies to make proteins which are responsible for strength of our muscles and for linking sarcomeres together to help in movement of body. Each gene associated with MFM provide instructions for proteins that perform slightly different function in muscle. So, the symptoms exhibited by any MFM patient depend on the gene altered. In about 50 % of people, the exact genetic cause of the disease is not identified. This indicates that there are other genes related to MFM which are not yet discovered.
Based on the gene involved, some forms of MFM are:
- Desminopathy
- Alpha-B crystallinopathy
- Myotilinopathy
- Zaspopathy
- Filaminopathy
- BAG3-opathy
Inheritance of Myofibrillar Myopathies
Myofibrillar myopathy can be inherited in different ways depending on the exact gene that is changed. In most cases, MFM are caused by change in genes that are inherited in an autosomal dominant manner. MFM can exhibit reduced penetrance and variable expressivity. Reduced penetrance means that some people who have genetic changes that cause MFM may not show any signs or symptoms of the MFM but can still pass it to their children. Variable expressivity means that the exact symptoms of each person can differ, even within the same family. For example, some affected individuals may have both cardiomyopathy and muscle weakness, while others may just have one or the other.
If MFM is caused by a change in the FHL1 gene, it is inherited in an X-linked manner. The FHL1 gene is located on the X chromosome. The X chromosome is one of the sex chromosomes. Each woman has two X chromosomes, and each man has one X chromosome and one Y chromosome. Men have only one X chromosome, so they only have one copy of the FHL1 gene. If this gene of men has a mutation, they will have MFM. However, women have 2 X chromosomes. If there is mutation in this gene in woman, there is still another copy of working FHL1. So, they don’t show any signs and symptoms of the disease but are carriers of the disease.
Diagnosis of MFM
The generic diagnosis of MFM rests on the following;
- History of slow progressive weakness. In some individual, it is accompanied by muscle atrophy, stiffness and aching and paresthesia. Physical examination reveals proximal and distal weakness. Facial weakness is uncommon but can occur.
- Electromyography (EMG) can be used to diagnose abnormal electrical irritability (positive sharp waves, myotonic discharges). In about 20 % of individuals, abnormal nerve conduction is detected.
- Muscle histology that reveals small vacuoles, decreased oxidative enzyme activity in abnormal fiber regions and alterations in hyaline and granular material in muscle fiber.
- Electron microscopy of muscle reveals myofibrillar degeneration commencing in Z fibers, disintegration of sarcomeres.
- Immunocytochemical studies of muscles that shows abnormal ectopic expression of desmin (75%), myotilin (90% of abnormal fibers), α-B crystallin (75%), dystrophin (70%), and β-amyloid precursor protein (70%).
- Serum creatine kinase concentration can be normal or elevated (up to 15-fold more than normal level)
- Other tissues, Myocardial biopsies show desmin-immunoreactive cytoplasmic inclusions, especially near intercalated disks and interstitial fibrosis. Peripheral nerve biopsies, in a few descriptions, show accumulation of neurofilaments, neurotubules, and formation of axonal spheroids.
The diagnosis of specific MFM is based on molecular genetic testing.
Treatment
The treatment depends on the signs and symptoms present in each person. Patient who have progressive muscle weakness may require physical therapies and assistive devices such as wheelchair or cane. Some patients who shows cardiomyopathy or arrythmia may improve with a pacemaker or ICD (Implantable Cardioverter Defibrillator). In severe cases, some patients may require heart transplant. In some individuals, progressive muscle weakness may affect ability of lung muscles to work, especially at night. They may require respiratory support such as ventilator. Regular monitoring of heart, lungs and muscles are recommended.
There is no single best treatment for MFM as the condition varies among individuals. If anyone had been diagnosed with this condition, obtaining genetic counselling and learning how it may affect own’s offspring is the best idea.
References
- Selcen D, Engel AG. Mutations in myotilin cause myofibrillar myopathy. Neurology. 2004; 62: 1363–71.
- Selcen D, Bromberg MB, Chin SS, Engel AG. Reducing bodies and myofibrillar myopathy features in FHL1 muscular dystrophy. 2011; 77: 1951–9.
- Fichna JP, Maruszak A, Żekanowski C. Myofibrillar myopathy in the genomic context. J Appl Genet. 2018 Nov; 59(4): 431-439.
- Olivé M, Kley RA, and Goldfarb LG. Myofibrillar myopathies: new developments. Current Opinion in Neurology. October 2013; (26)5: 527-535.
- Montse Olivé, Rudolf A. Kley, and Lev G. Goldfarb. Myofibrillar myopathies: new developments. Rev Neurol (Paris). 2016 Oct; 172(10): 594-606.
- Sabrina Batonnet-Pichon, Anthony Behin, Eva Cabet, Florence Delort, Patrick Vicart, and Alain Lilienbaum. Myofibrillar Myopathies: New Perspectives from Animal Models to Potential Therapeutic Approaches. J Neuromuscul Dis. 2017; 4(1): 1–15.
- Gregory J. Latham1 & Grace Lopez. Anesthetic considerations in myofibrillar myopathy. Pediatric Anesthesia. 25 (2015): 231–238.